Drug Level & Toxicity Simulator
Physiological Variable Impact Tool
Select a body condition to see how it shifts a drug from the 'Helpful Zone' to the 'Toxic Zone'.
Simulation Output
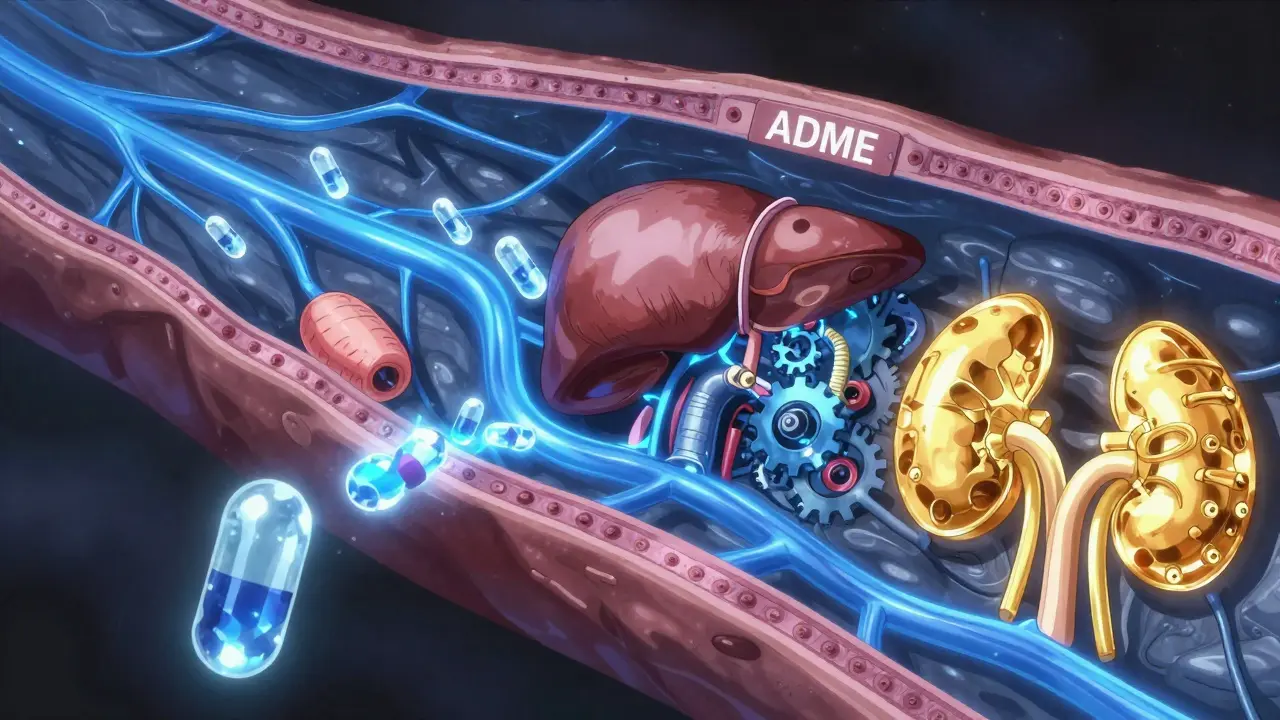
When you swallow a pill or get an injection, your body immediately starts a complex four-stage journey known as the ADME process. If any part of this chain breaks down-maybe your kidneys are sluggish or your liver is processing things too slowly-the drug can build up to toxic levels in your bloodstream. This is often when the "side effects" we complain about actually turn into dangerous adverse reactions.
The ADME Journey: How Drugs Move Through You
To understand side effects, we first have to look at the four stages of drug movement. Think of it as a logistics chain; if there is a bottleneck at any stage, the whole system is affected.
First is Absorption. This is how the drug gets from the point of entry into your blood. If you take a pill, it has to survive your stomach acid and cross the intestinal wall. Not everything makes it; this is called bioavailability. For instance, oral drugs often only hit 40-60% bioavailability because of the "first-pass effect," where the liver grabs a chunk of the drug before it ever reaches the rest of your body. Intravenous (IV) drugs, on the other hand, have 100% bioavailability because they skip the line and go straight into the vein.
Next comes Distribution. Once in the blood, the drug needs to get to the target site. Some drugs stay mostly in the plasma, while others, like certain antidepressants, dive deep into fatty tissues. A huge factor here is protein binding. Imagine albumin, a protein in your blood, acting like a sponge. If 98% of a drug like warfarin is stuck to albumin, only that tiny 2% left floating is actually active. If something knocks the drug off that protein, you suddenly have way more active medication in your system, which can lead to internal bleeding.
Then we hit Metabolism. This happens mostly in the liver using Cytochrome P450 enzymes (or CYP enzymes). Their job is to turn the drug into a water-soluble form so you can pee it out. One specific enzyme, CYP3A4, is a powerhouse-it handles about 50% of all clinically used drugs.
Finally, there is Excretion. Your kidneys are the primary exit door. They filter the blood and send the waste to your bladder. If your glomerular filtration rate (GFR)-the speed at which your kidneys filter blood-drops, the drug lingers longer, increasing the risk of toxicity.
Why Pharmacokinetics Leads to Side Effects
Side effects aren't always random. They are often the direct result of a pharmacokinetic "mismatch." When the concentration of a drug in your blood exceeds the therapeutic window, you move from the "helpful" zone into the "toxic" zone.
Take phenytoin, used for seizures. If the level is around 10 mcg/mL, it works great. But if it climbs above 20 mcg/mL, toxicity rates jump from 2% to 30%. This usually happens because the body can't excrete the drug as fast as it's being put in.
Another culprit is active metabolites. Sometimes the liver doesn't just deactivate a drug; it turns it into a new, active version. Diazepam is a classic example. Its metabolite, desmethyldiazepam, has a half-life of up to 100 hours. In older adults, whose livers and kidneys move slower, this metabolite piles up, leading to prolonged sedation and a much higher risk of falls.
Genetic differences also play a massive role. Some people are "poor metabolizers" due to their DNA. For example, about 3-10% of Caucasians have a variation in the CYP2D6 enzyme. If these people take codeine, their body can't convert it into morphine effectively, meaning the drug doesn't work. On the flip side, other genetic variants can make warfarin five times more likely to cause bleeding if the dose isn't lowered.

Drug-Drug Interactions and the CYP Bottleneck
When you take multiple medications, they often fight for the same "processing plant" in the liver. This is a common cause of unexpected side effects. If two drugs both need the CYP3A4 enzyme, one might block the other from being processed.
Consider a patient taking simvastatin for cholesterol who starts taking clarithromycin for an infection. Clarithromycin is a potent CYP3A4 inhibitor. It essentially shuts down the exit door for simvastatin, causing levels to spike 10-fold. This can lead to rhabdomyolysis-a severe breakdown of muscle tissue-increasing the risk from a tiny 0.04% to a dangerous 0.5%.
| Variable | Change in Body | Effect on Drug Level | Typical Side Effect Risk |
|---|---|---|---|
| Renal Function | Low GFR (Kidney Failure) | Increased accumulation | Toxicity / Organ damage |
| Liver Enzymes | CYP Inhibition (Drug interaction) | Slower metabolism | Overdose symptoms |
| Protein Binding | Low Albumin (Malnutrition) | More "free" active drug | Increased potency/bleeding |
| Age | Reduced Hepatic/Renal clearance | Prolonged half-life | Extreme sedation / Confusion |
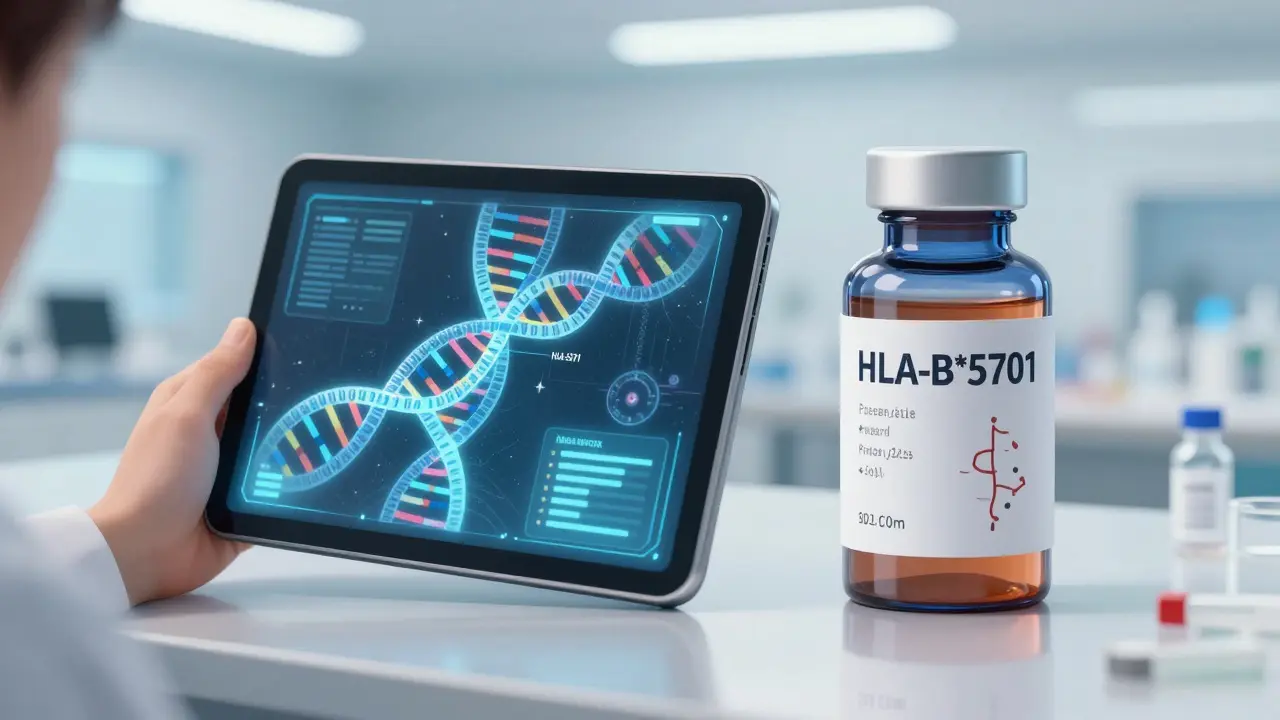
Personalizing the Dose: How We Fix the Problem
Because we all process drugs differently, the "one size fits all" dosing model is outdated. To prevent side effects, clinicians use Therapeutic Drug Monitoring (TDM). This involves drawing blood at very specific times-usually the "trough level," which is just before the next dose-to see exactly how much drug is left. TDM can reduce adverse events by 25-35% in complex cases.
We also use mathematical formulas. The Cockcroft-Gault equation helps doctors estimate kidney function based on your age and weight, allowing them to adjust the dose of powerful antibiotics like vancomycin. Without this, a patient with poor kidney function could easily suffer from nephrotoxicity, where the drug intended to kill bacteria ends up damaging the kidneys instead.
The future is moving toward precision medicine. We are now seeing the rise of pharmacogenetic testing. For example, testing for the HLA-B*5701 allele can prevent 90% of severe hypersensitivity reactions to the drug abacavir. AI platforms like DoseMeRx are also beginning to automate dosing, reducing errors in high-risk medications by over 60%.
Practical Tips for Managing Your Medications
Knowing how your body processes drugs helps you have better conversations with your doctor. You aren't just a passive recipient of a pill; your biology is actively interacting with the chemistry.
- Be honest about all supplements: Some herbal supplements (like St. John's Wort) are powerful CYP enzyme inductors. They can make your prescription drugs disappear from your system too quickly, making them ineffective.
- Mention your age and health history: If you have a history of kidney or liver issues, ensure your doctor knows. A dose that is safe for a 30-year-old might be toxic for a 70-year-old.
- Timing matters: Some drugs need a specific gastric pH to be absorbed. Taking a medication with an antacid might stop it from ever reaching your bloodstream.
- Watch for new symptoms: If you start a new med and feel a strange side effect, it might be a sign of a drug-drug interaction or a metabolic bottleneck.
What is the difference between pharmacokinetics and pharmacodynamics?
Pharmacokinetics is what your body does to the drug (absorption, distribution, metabolism, and excretion). Pharmacodynamics is what the drug does to your body (how it binds to receptors and creates a biological effect). In short: kinetics is the journey; dynamics is the action.
Why do some people experience more side effects than others?
It usually comes down to genetic variations in enzymes (like the CYP450 system) and organ function. For example, if your liver processes a drug slowly, it stays in your blood longer, increasing the likelihood of side effects. Age and kidney health also play huge roles in how quickly a drug is cleared from the body.
Can I change how my body processes a drug?
You cannot change your genetics, but you can influence your metabolic environment. Diet, hydration, and other medications can speed up or slow down enzymes in the liver. This is why doctors tell you to avoid grapefruit juice with certain statins, as it inhibits the enzyme responsible for breaking the drug down.
What is first-pass metabolism?
First-pass metabolism happens when a drug is absorbed from the gut and passes through the liver via the portal vein before reaching general circulation. The liver may metabolize a large portion of the drug, significantly reducing the amount that actually reaches the rest of the body.
How does kidney function affect drug dosing?
The kidneys are responsible for excreting many drugs. If your glomerular filtration rate (GFR) is low, the drug isn't cleared as efficiently. This leads to higher concentrations in the blood, which can cause toxicity unless the dose is lowered or the interval between doses is increased.
15 Comments
Write a comment
Recent-posts

Nov, 23 2025

Aug, 13 2025

Dec, 30 2025

Dec, 11 2025

Mar, 26 2026
Categories
Tags
- online pharmacy
- drug interactions
- side effects
- generic drugs
- online pharmacy UK
- medication safety
- GoodRx coupons
- drug safety
- opioid side effects
- medication timing
- pill organizer
- generic medications
- medication adherence
- safe online pharmacy
- Tadalafil
- arthritis medication
- buy medication online
- pharmacy safety
- prescription medication
- quit smoking



Doug DeMarco
April 11, 2026 AT 23:56This is such a great breakdown of how the body works! It's always wild how a tiny genetic switch in the liver can totally change how a med hits you. Stay curious everyone! 😊✨
danny Gaming
April 13, 2026 AT 12:16basic science stuff... we got better meds in US anyway probly just overcomplicating it for ppl who cant read a label properly lol
Trey Kauffman
April 14, 2026 AT 21:10Oh sure, because assuming we're all just biological logistics chains is a wonderful way to view the human experience. Truly profound.
Robin Walton
April 14, 2026 AT 23:45I really appreciate the part about older adults and the half-life of medications. It explains so much about why my grandparents struggle with certain prescriptions. It's so important to be patient with them.
Ben hogan
April 16, 2026 AT 14:11The reductionist nature of this post is honestly exhausting. To suggest that AI and a few equations can capture the nuance of human biochemistry is the height of arrogance and intellectual laziness. I've read textbooks on CYP450 that make this look like a children's nursery rhyme, and frankly, the lack of depth here is an insult to anyone with a basic understanding of organic chemistry. It's a shallow summary for people who prefer bullet points over actual rigorous study. Pathetic.
Danny Wilks
April 17, 2026 AT 23:59It is quite fascinating to consider how these chemical interactions mirror the diverse ways different cultures approach wellness, although the clinical precision of the ADME process provides a universal framework that transcends regional variations in medical practice. I find the concept of bioavailability particularly intriguing because it highlights the inherent inefficiency of the human digestive system when faced with synthetic compounds.
Kelly DeVries
April 19, 2026 AT 15:01omg i literally had a reaction to a supplement once and didnt even think about the CYP enzymes lol this is why you gotta be careful with what you put in your body honestly
Suchita Jain
April 21, 2026 AT 10:53It is imperative that one maintains a strict regimen of medical consultation before altering any medication based on such a generalized text. One must adhere to the guidance of a certified professional to ensure safety.
Simon Stockdale
April 22, 2026 AT 01:14my uncle took some weird herbal tea and it messed with his blood pressure meds big time and he was all dizzy as heck and it just shows that these big pharma companies don't even tell us half the stuff about how our own bodies react while they just make a killing off us in the states and we just gotta deal with the side effects like we're lab rats in a giant experiment!
emmanuel okafor
April 23, 2026 AT 06:29nature and science are just two ways of saying the same thing in the end
kalpana Nepal
April 25, 2026 AT 00:50This knowledge is good but we must remember that our traditional ways are also strong. Science is just a tool to explain what we already know about the body.
Thabo Leshoro
April 26, 2026 AT 18:39The pharmacokinetics here... it's really complex!!! The GFR is such a vital marker... for renal clearance... it's scary how it works!!!
Chad Miller
April 27, 2026 AT 18:42who cares about the math just give me the med that works and stop with the jargon
Ryan Hogg
April 28, 2026 AT 19:14I've spent years feeling like a freak because my meds never worked and reading this just makes me feel more broken. Like, why do I have to be the 3% that doesn't process things right? It's just another thing to deal with in a life already full of medical failures.
Lynn Bowen
April 30, 2026 AT 14:51Interesting to see how this varies across different healthcare systems globally.